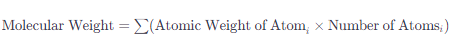💡pubchem이란? 미국 환경보호청(EPA)에서 개발한 분산 구조 검색 가능 독성(DSSTox) 데이터베이스이다. 이 데이터베이스는 화학물질의 잠재적 건강 영향에 대한 정보를 제공함으로써 연구 및 규제 결정을 지원하도록 설계되었고 사용자들은 화학물질의 구조, 이름, 또는 다른 특성을 기반으로 검색하고, 화학물질의 위험, 노출 위험 및 환경 영향과 같은 독성 데이터를 알아볼 수 있는 사이트이다.
💡smiles식이란? SMILES(Simplified Molecular Input Line Entry System)는 화학 물질의 구조를 문자열로 표현하는 방식이다. 이 시스템은 화학자들이 컴퓨터와 데이터베이스에서 화학 물질을 쉽게 저장, 검색 및 교환할 수 있도록 설계되었다. SMILES 표기법은 원자와 결합을 문자와 숫자로 나타내며, 이를 통해 복잡한 화학 구조도 간단한 문자열로 표현할 수 있다.
링크 접속 한 홈페이지 화면
Daily dose data에 대한 data table
데이터 분석
데이터 테이블을 잠시 살펴보자면 해당 csv파일에는 Tested Substance가 smiles식으로 표현된 식, 활성/비활성 상태인지 나타내는 Activity, 이 약물에 대한 하루 섭취 권장량인 Dose_MRDD_mg, 하루 섭취 권장량을 분자량으로 나눈 Dose_MRDD_mmol등이 있었다.
이후 데이터를 다운 받고 분석을 하기 전 데이터의 분포를 보기 위해 EDA를 수행하였다.
import pandas as pd
# 파일 읽기
daily_dose_data = pd.read_csv('AID_1195_datatable.csv')
daily_dose_data.head(5)
# 데이터 column 확인
daily_dose_data.info()
# 결측치 확인
daily_dose_data.isnull().sum()
확인 결과 총 11개의 열이 있었고 결측치까지 확인한 결과 결측치가 존재하였기 떄문에 이를 제거하는 작업을 진행하였다.
MolecularWeight(분자량), LogP값 계산을 통해 데이터의 분포도를 확인하고자 이 두 개의 열을 계산한 후 추가하였다.
💡MolecularWeight이란? 화학 물질을 구성하는 원자의 상대적 질량의 합이다. 단위는 일반적으로 그램/몰(g/mol)로 표현되며, 이는 한 몰의 해당 화합물이 가지는 질량을 의미한다. 💡LogP란? 약물의 생체 내 흡수, 분포, 대사, 배설(ADME) 특성을 평가하는 데 중요한 파라미터로 사용된다. 예를 들어, 약물이 세포막을 통과하여 효과적으로 작용하기 위해서는 적절한 친수성과 소수성의 균형이 필요하며, 이는 LogP 값으로 평가할 수 있다. LogP 값이 너무 높으면 약물이 지나치게 소수성이 되어 수용성이 떨어지고, 너무 낮으면 세포막을 통과하는 능력이 떨어질 수 있다.
## smile식으로 molecular weight 계산
from rdkit import Chem
from rdkit.Chem import Descriptors
from rdkit.Chem import Crippen
# MolecularWeight 열 추가
daily_dose_data['MolecularWeight'] = daily_dose_data['PUBCHEM_EXT_DATASOURCE_SMILES'].apply(lambda x: Chem.MolFromSmiles(x) if pd.notna(x) else None)
daily_dose_data['MolecularWeight'] = daily_dose_data['MolecularWeight'].apply(lambda mol: Descriptors.MolWt(mol) if mol is not None else None)
# LogP 열 추가
daily_dose_data['LogP'] = daily_dose_data['PUBCHEM_EXT_DATASOURCE_SMILES'].apply(lambda x: Crippen.MolLogP(Chem.MolFromSmiles(x)) if pd.notna(x) else None)
print(daily_dose_data[['MolecularWeight', 'LogP']])
이후 분포를 보기 위해 train, test셋으로 나누고 Dose_MRDD_mmol에 대한 분포를 살펴보았다.
대체적으로 train, test가 고르게 분포되어있었고 타겟 변수(Dose_MRDD_mmol)에 log scale한 이유는 해당 값이 1e-8과 같이 너무 작아 분포를 좀 더 고르게 보기 위해 적용하였다.
데이터 분포는 bell curve(정규분포곡선) 모양을 따르는 것을 보아 이를 통해 이 데이터는 회귀분석에 적절하다는 것으로 판단되었다.
import matplotlib.pyplot as plt
from sklearn.model_selection import train_test_split
import numpy as np
# train, test 나누기
train_data, test_data = train_test_split(daily_dose_data, test_size=0.2, random_state=42)
# train, test 결합
combined_data = pd.concat([train_data, test_data])
## 산점도(train vs test)
plt.figure(figsize=(10, 6))
plt.scatter(train_data['MolecularWeight'], train_data['LogP'], color='b', marker='o', alpha=0.7, label='Training Set')
plt.scatter(test_data['MolecularWeight'], test_data['LogP'], color='r', marker='o', alpha=0.7, label='Testing Set')
plt.title('Scatter Plot of Molecular Weight vs. LogP (Combined Set)')
plt.xlabel('Molecular Weight')
plt.ylabel('LogP')
plt.legend()
plt.show()
## 히스토그램(train vs test)
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(15, 6))
# Training Set
ax1.hist(np.log10(train_data['Dose_MRDD_mmol']), bins=100, color='b', alpha=0.7, label='Training Set')
ax1.set_title('Histogram of Dose_MRDD_mmol (Training Set)')
ax1.set_xlabel('Dose_MRDD_mmol')
ax1.set_ylabel('Frequency')
ax1.legend()
# Testing Set
ax2.hist(np.log10(test_data['Dose_MRDD_mmol']), bins=100, color='r', alpha=0.7, label='Testing Set')
ax2.set_title('Histogram of Dose_MRDD_mmol (Testing Set)')
ax2.set_xlabel('Dose_MRDD_mmol')
ax2.set_ylabel('Frequency')
ax2.legend()
# 그래프 출력
plt.tight_layout()
plt.show()
train, test으로 나눈 데이터를 drop_duplicate를 이용해 중복 값을 제거 한 후 csv파일로 저장했다.
# train set 중복 제거 후 csv파일로 저장
train_data['Dose_MRDD_mmol'].value_counts()
train_data.drop_duplicates(subset=['Dose_MRDD_mmol'], inplace=True)
train_data.to_csv('train_data.csv')
# Test set 중복 제거 후 csv파일로 저장
test_data['Dose_MRDD_mmol'].value_counts()
test_data.drop_duplicates(subset=['Dose_MRDD_mmol'], inplace=True)
test_data.to_csv('test_data.csv')